We combine structural biology, biochemical, and biophysical approaches to study the structure and mechanism of transmembrane transport proteins. We aim to reveal the molecular basis for their physiological and pathophysiological functions.

We combine structural biology, biochemical, and biophysical approaches to study the structure and mechanism of transmembrane transport proteins. We aim to reveal the molecular basis for their physiological and pathophysiological functions.
Voltage-dependent ion channels are membrane proteins that facilitate the rapid conduction of ions, regulated by the voltage across the membrane. In electrically excitable cells, such as neurons and cardiac myocytes, ion channels -- including voltage-gated sodium (Nav) channels and voltage-gated calcium (Cav) channels -- play a crucial role in the swift propagation of electrical signals, driven by the generation of action potentials. The voltage sensor, a region of the protein containing charged amino acids, undergoes relocation in response to changes in the membrane's electric field. This movement initiates a conformational change in the gate of the conductive pathway, thereby regulating the flow of ions. Nav and Cav channels are significant targets for drug development. Our lab aims to investigate the working mechanism and structural pharmacology of Nav and Cav channels rhrough structural biology, molecular biology, electrophysiology and cell biology techniques.
Nine isoforms of Nav channels are present in the human body, with distinct distributions across various tissues and organs. Dysfunctions or mutations in these channels can lead to severe conditions, including epilepsy, arrhythmia, and erythromelalgia. Furthermore, Nav channels are critical targets for clinical anesthetics and disease treatment. Therefore, we aim to elucidate the structure and working mechanism of Nav channels, which hold significant physiological relevance and are valuable for basic research. These channels represent ares of active investigation in pharmacology within the life sciences.
1. Structures of the first eukaryotic voltage-gated sodium channel NavPaS:
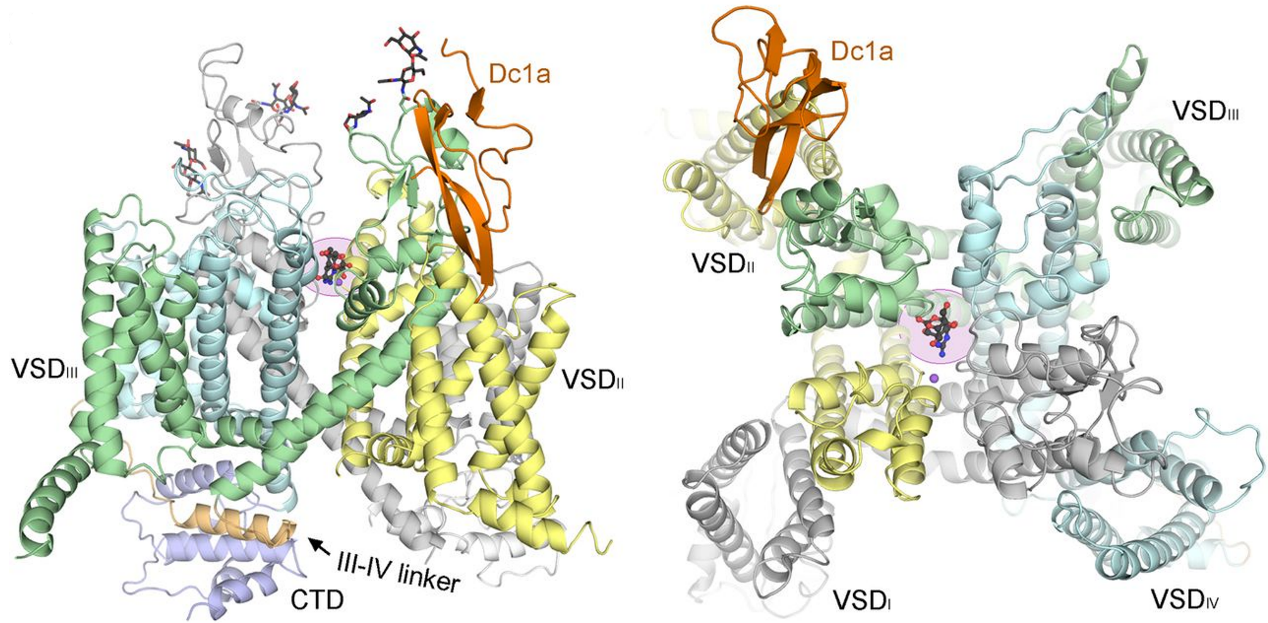
On Feb 9, 2017, we reported the cryo-EM structure of the eukaryotic voltage-gated sodium channel, NavPaS, at a resolution of 3.8 Å. It was the first eukaryotic Nav channel structure resolved at near-atomic resolution, and has laid the foundation for understanding its functional mechanisms and the pathogenesis of related diseases. The near-atomic structure provides the detailed architecture of NavPaS, allowing for side-chain assignments across the complete transmembrane fold, extracellular loops of the pore domain, the intact III-IV linker, and the carboxy-terminal domain (CTD). Animal toxins targeting on Nav channels can be broadly classified into two categories: pore blockers and gating modifiers. Thetetrodotoxin (TTX) and saxitoxin (STX) are classic pore blockers responsible for pufferfish and shellfish poisoning in humans, respectively. We reported the structures of the NavPaS bound to the gating modifier toxin Dc1a, both alone or in complex with TTX or STX, at resolutions of 2.8 Å, 2.6 Å and 3.2 Å, respectively. These structures reveal the molecular details of toxin binding and provide insights for the structure-based development of Nav channel drugs. [Science 355, eaal4326 (2017)] [Science 362, eaau2596 (2018)]
2. Structural analysis of Nav1.1 reveals mutational hotspots for sodium channelopathies
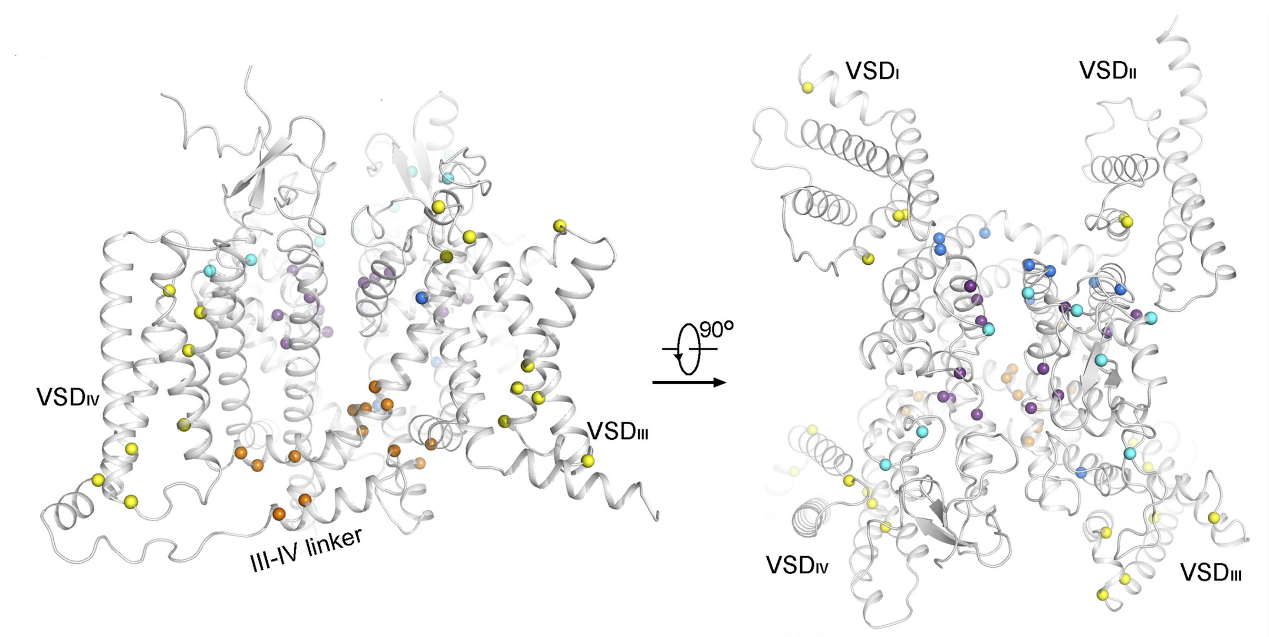
Nav1.1, encoded by SCN1A in humans, is a major subtype of Nav channels in the brain. Up to 900 nonsense and missense mutations in SCN1A have been identified in patients with epilepsy syndromes. We determined the cryo-EM structure of the human Nav1.1–β4 complex at a resolution of 3.3 Å. Comparative structural analysis of hundreds of missense disease mutations in Nav1.1 and Nav1.5-E1784K reveals 70 loci common to these two channels. Several clusters, defined as mutational hotspots, were identified and generally classified into structural mutations and functional mutations. These comparative structural analyses establish a framework for dissecting the structure–function relationship of Nav disease variants and will facilitate the development of precision medicine for various sodium channelopathies. [PNAS 118, e2100066118 (2021)]
3. Molecular basis for pore blockade of human Nav1.2 by the μ-conotoxin KIIIA
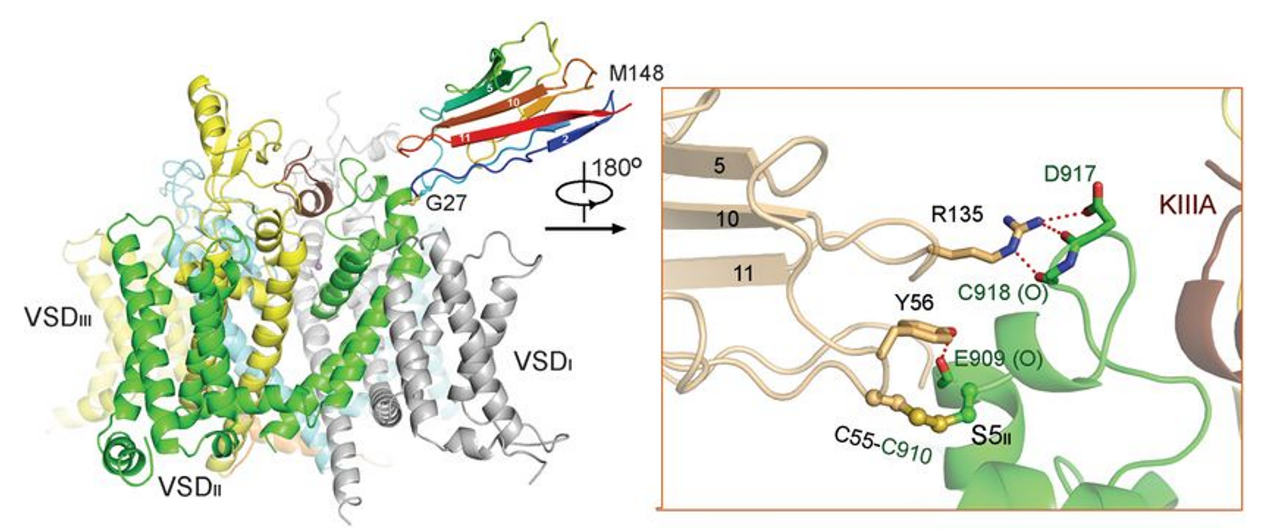
Nav1.2, encoded by SCN2A, is responsible for initiating and propagating action potentials in the central nervous system. We reported the cryo–EM structure of human Nav1.2 bound to the peptidic pore blocker, the μ-conotoxin KIIIA, in the presence of an auxiliary subunit, β2, at an overall resolution of 3.0 Å. The immunoglobulin domain of β2 interacts with the shoulder of the pore domain through a disulfide bond. The 16-residue KIIIA interacts with the extracellular segments in repeats I to III, placing Lys7 at the entrance of the selectivity filter. Many interacting residues are specific to Nav1.2, revealing a molecular basis for the specificity of KIIIA. This structure establishes a framework for the rational design of subtype-specific blockers for Nav channels. [Science 363, 1309-1313 (2019)]
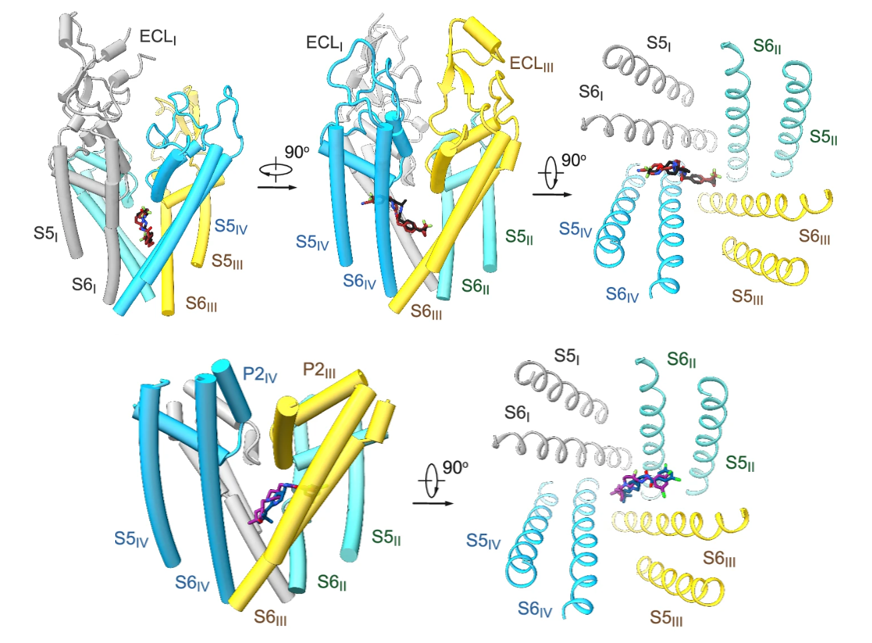
4. Structures of electric eel and finally human Nav1.4
Nav1.4, encoded by SCN4A in humans, functions in skeletal muscle. We firstly determined the cryo-EM structure of EeNav1.4, the Nav channel from electric eel, in complex with the β1 subunit at a resolution of 4.0 Å. With extensive effort, we subsequently solved the cryo–EM structure of the human Nav1.4 (hNav1.4)-β1 complex at a resolution of 3.2 Å. These two structures were nearly identical. Accurate model building of hNav1.4-β1 was done for the pore domain, the voltage-sensing domains, the fast inactivation motif, and the β1 subunit, providing insight into the molecular basis for Na+ permeation and the kinetic asymmetry of the four repeats. Structural analysis of reported functional residues and disease mutations corroborates an allosteric blocking mechanism for the fast inactivation of Nav channels. [Cell 170, 470-482 (2017), Science 362, eaau2486 (2018)]
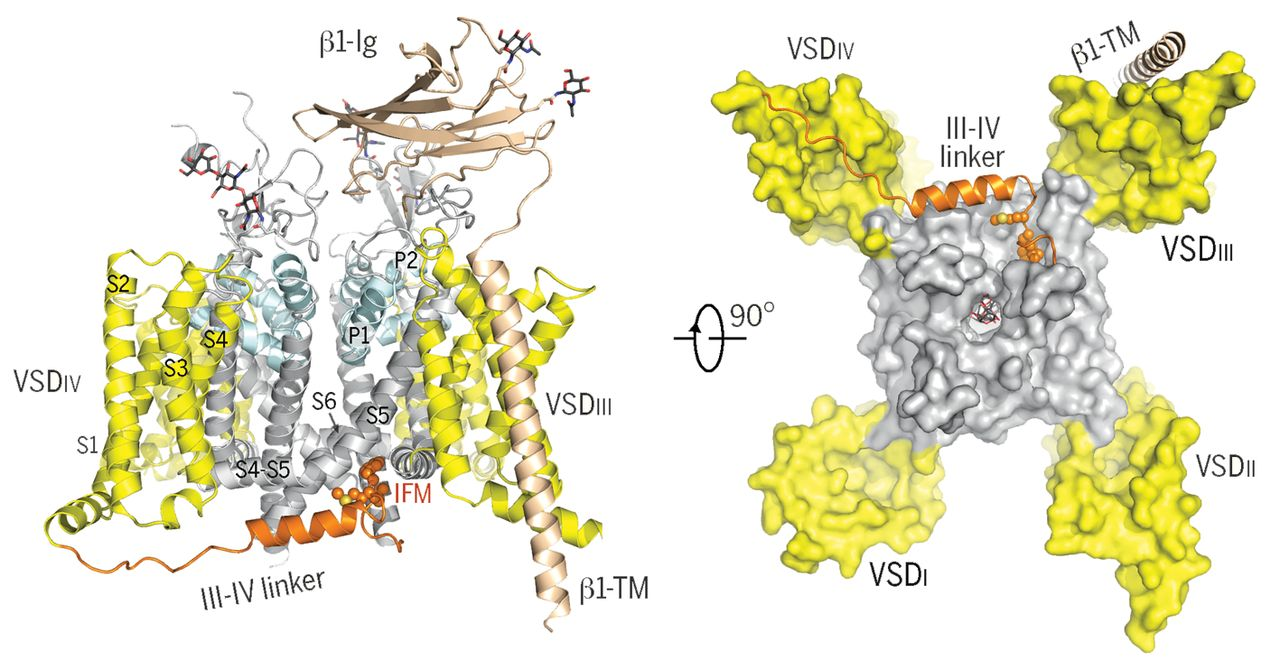
5. Structural investigation of cardiac voltage-gated sodium channel Nav1.5
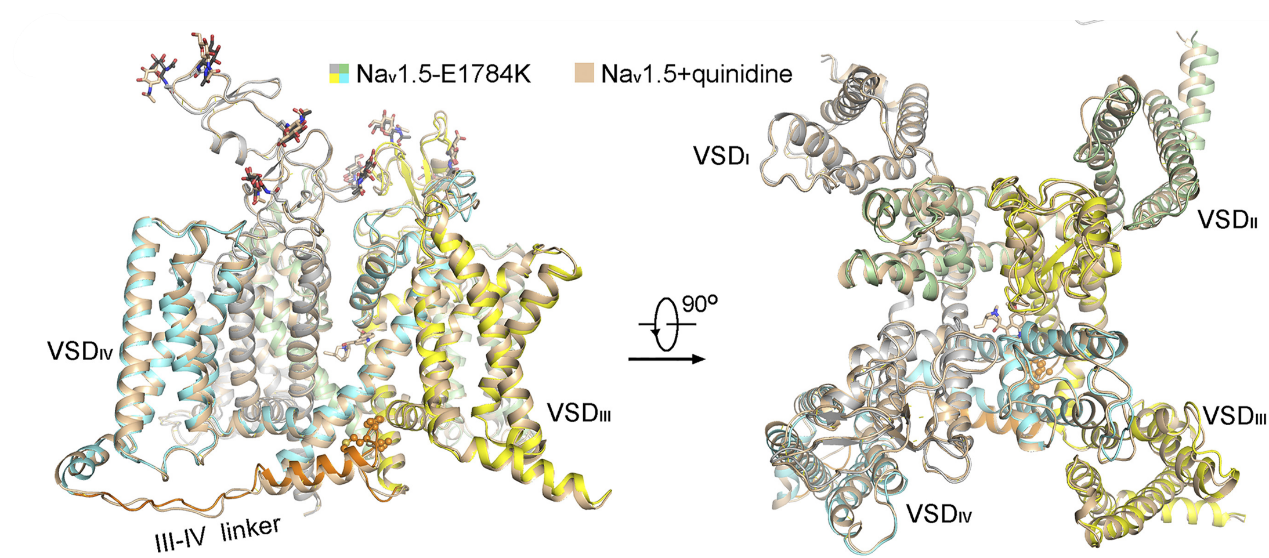
Nav1.5, encoded by SCN5A, is the primary Nav channel in the heart. Dysfunction of Nav1.5 is associated with several arrhythmia syndromes, exemplified by type 3 long QT syndrome (LQT3) and Brugada syndrome (BrS). We first determined the cryo-EM structure of Nav1.5 bound to quinidine at a resolution of 3.3 Å, revealing the molecular basis for the mechanism of action of Class Ia antiarrhythmic drugs. Immediately thereafter, we reported the cryo-EM structure of Nav1.5-E1784K, the most common variant associated with both LQT3 and BrS. From this structural analysis, we proposed a “door wedge” model to elucidate the mechanism of fast inactivation. Together with previous studies, the establishment of the structure-function relationship and understanding of the disease variants will facilitate the development of antiarrhythmic drugs. [Angew. Chem. Int. Ed. Engl. 60, 11474-11480 (2021)] [PNAS 118, e2100069118 (2021)]
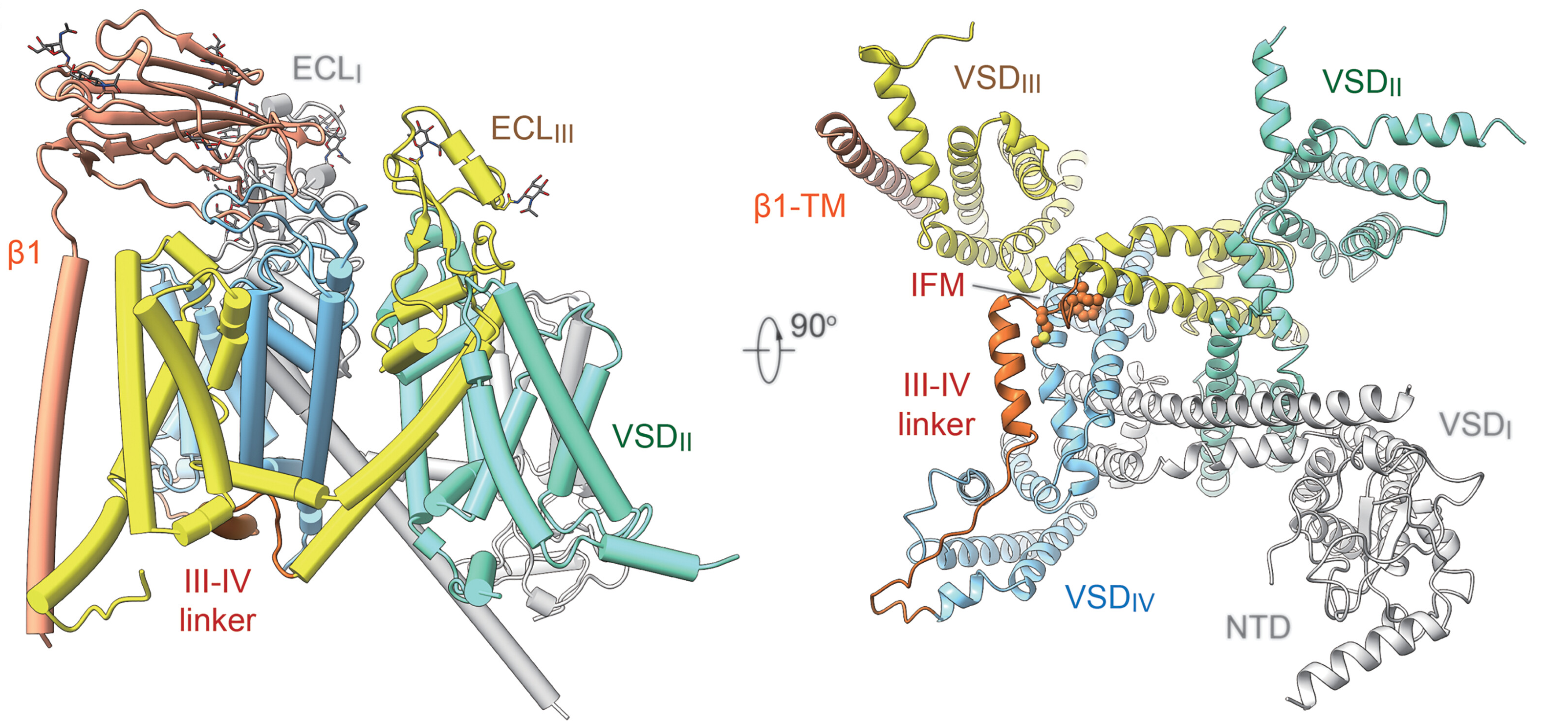
6. Cryo-EM structure of human voltage-gated sodium channel Nav1.6
Nav1.6, encoded by SCN8A, is predominantly expressed in the central nervous system (CNS) and plays a vital role in neuronal firing. Abnormal activity of Nav1.6 is linked to neurological disorders, such as epilepsy. A high-resolution cryo-EM structure of human Nav1.6 co-expressed with β1 and FHF2B has been reported by our group, in which an intact extracellular loop above the pore domain and a longer glycan chain in the first repeat are resolved. A previously uncharacterized density associated with a yet-to-be-identified molecule provides clues to the development of antiepileptic drugs specifically targeting Nav1.6. [PNAS 120, e2220578120 (2023)]
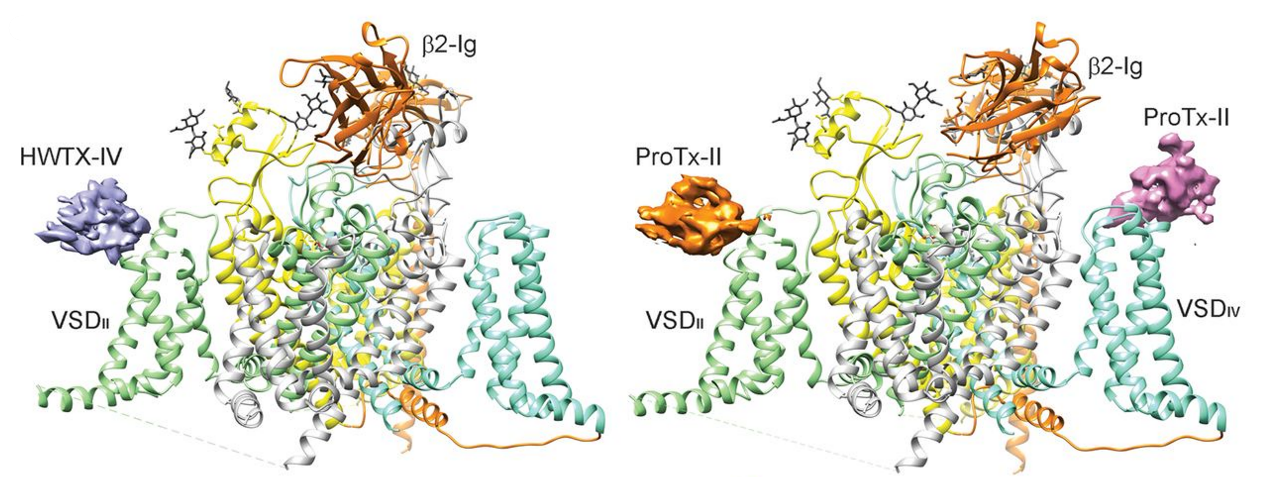
7. Structural investigation of pain-related voltage-gated sodium channel Nav1.7
Nav1.7, encoded by SCN9A in humans, is primarily located in the dorsal root ganglia and plays crucial roles in pain transduction. Due to its physiological importance, Nav1.7 has gained recognition as a potential target for developing non-addictive analgesics.
We determined the cryo–EM structures of the human Nav1.7-β1-β2 complex bound to two combinations of pore blockers and gating modifier toxins (GMTs): tetrodotoxin with protoxin-II and saxitoxin with huwentoxin-IV, both at overall resolutions of 3.2 Å. These structures illuminate the mechanistic understanding of Nav1.7 function and its relationship with diseases, providing a foundation for structure-aided development of analgesics.
The structures of Nav1.7 captured in different conformations will reveal its working mechanism and facilitate drug discovery. We rationally designed a Nav1.7 variant, Nav1.7-M11, which may be trapped in the closed-state inactivation conformation. Structural analysis of Nav1.7-M11 reveals VSDI in the fully down conformation, VSDII at an intermediate state, and the pore domain tightly closed. Structural comparison of Nav1.7-M11 with the WT channel provides unprecedented insight into the details of electromechanical coupling and offers a mechanistic interpretation for various pain-related mutations. [Science 363, 1303-1308 (2019), Cell Rep 39, 110735 (2022), PNAS 119, e2209164119 (2022), Nat. Commun. 14, 3224 (2023)]
8. Structural basis for high-voltage activation and subtype-specific inhibition of human Nav1.8
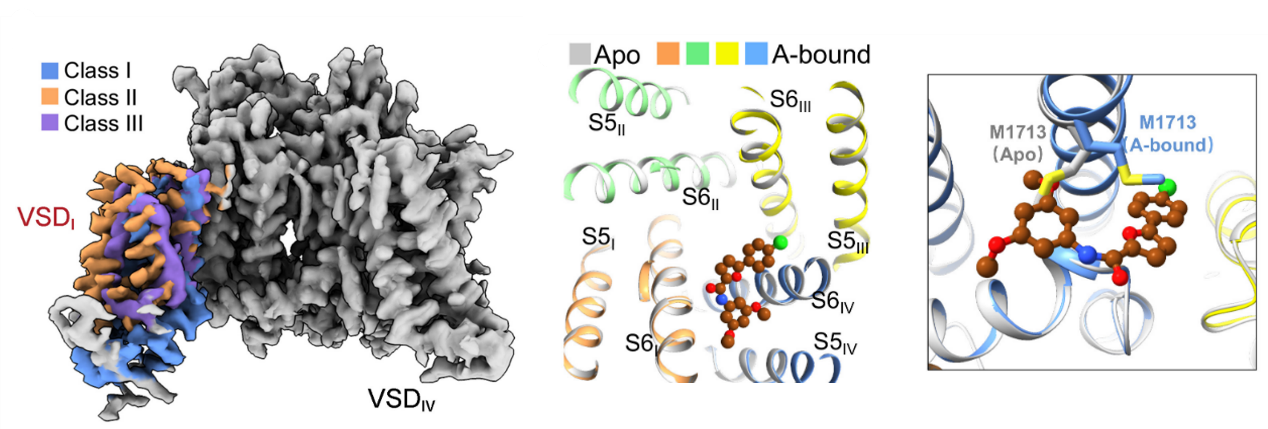
Nav1.8, a tetrodotoxin (TTX)-resistant subtype encoded by SCN10A, is primarily expressed in sensory neurons, particularly in the dorsal root ganglia (DRG) neurons. Nav1.8 possesses several unique biophysical properties, such as activation at more depolarized voltages and slower inactivation with persistent current, which contribute to the hyperexcitability of DRG neurons. Additionally, Nav1.8 represents a potential target for developing non-addictive analgesics.
Cryo-EM structures of human Nav1.8 alone and bound to A-803467, a selective pore blocker, have been reported. Different conformations of VSDI were observed in Nav1.8. An extracellular interface between VSDI and the pore domain was determined, highlighting its dependence on higher voltage for activation. A-803467 clenched S6IV within the central cavity. Unexpectedly, channel selectivity for A-803467 was determined by non-ligand coordinating residues through an allosteric mechanism. [PNAS 119, e2208211119 (2022)]
Voltage-gated calcium (Cav) channels activate upon membrane depolarization, mediating Ca2+ influx to initiate various physiological events, including muscle contraction, neurotransmitter secretion, and regulation of gene expression. In mammals, there are ten members of Cav channels, which can be divided into three subfamilies (Cav1.1-1.4, Cav2.1-2.3, Cav3.1-3.3). Different subtypes of Cav channels mediate Ca2+ currents in various cell types, revealing different genetic, molecular and physiological properties. Additionally, Cav channels serve as primary drug targets for clinical treatment. Therefore, our lab aims to combine structural biology with biochemical and biophysical approaches to elucidate the molecular mechanisms of Cav channels, which is valuable for both basic research and pharmacological studies.
1. Structures of rabbit voltage-gated calcium channel Cav1.1 at near-atomic resolution:
The L-type voltage-gated calcium channel Cav1.1 is specialized for excitation-contraction coupling (ECC), which causes the contraction of skeletal muscle in response to changes in membrane potential. The Cav1.1 complex consists of the pore-forming α1 subunit and auxiliary subunits, including α2δ, β and γ subunits. The cryo-EM structures of rabbit Cav1.1 (rCav1.1) reveal the detailed architecture of a pseudo-tetrameric α1 subunit in complex with auxiliary subunits. Cav1.1 can also be modulated by various compounds, such as 1,4-dihydropyridine (DHP), benzothiazepine (BTZ). We reported the high-resolution structures of rCav1.1 in complex with nifedipine, diltiazem, verapamil and Bay-K 8644. These structures elucidate the modes of action of different Cav ligands and establish a framework for structure-guided drug discovery. [Science 350, aad2395 (2015), Nature 537, 191-196 (2016), Cell 177, 1495-1506 (2019)]
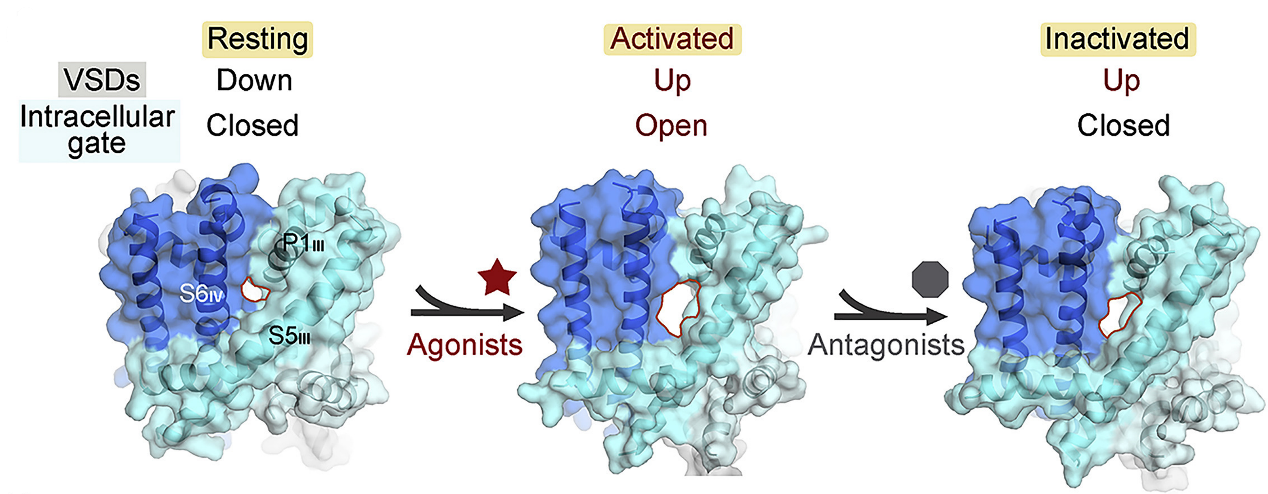
2. Structural basis for the severe adverse interaction of SOF and AMIO on L-type Cav channels:
Drug-drug interaction (DDI) between the antiviral sofosbuvir (SOF) and the antiarrhythmics amiodarone (AMIO) has been reported to cause fatal slowing of the heartbeat. We present a systematic study of Cav1.1 and Cav1.3 treated with AMIO or SOF alone, or SOF/MNI-1 (analog of SOF) combined with AMIO. Through structural analysis, we found that AMIO alone occupies the DHP site, while SOF alone is not found in the channel. However, in the presence of AMIO, SOF/MNI-1 is anchored in the central cavity of the pore, directly blocking the ion permeation pathway. Our study reveals the molecular basis for the physical and pharmacodynamics interaction of DDI in Cav channels. [Cell 185, 4801-4810 (2022)]
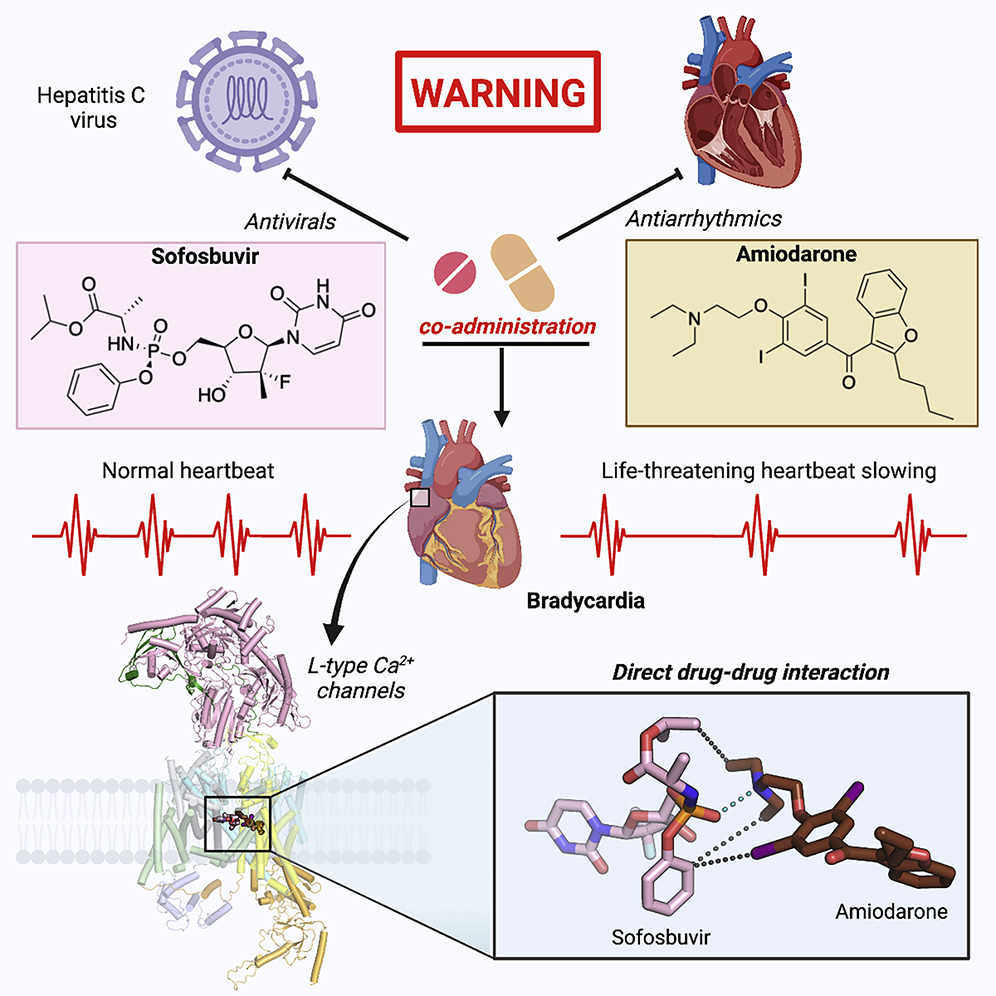
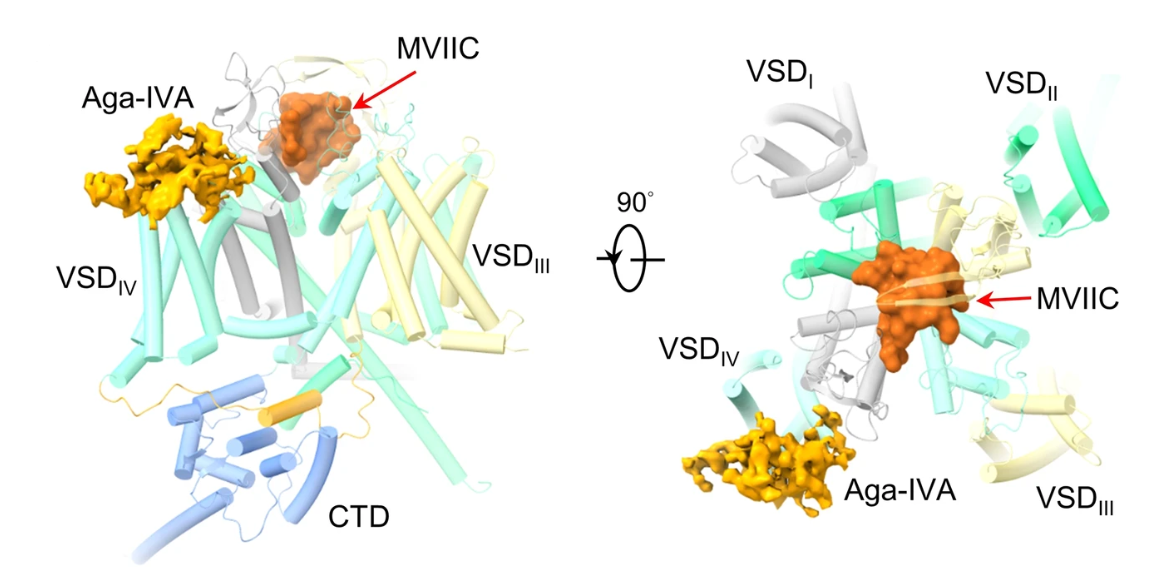
3. Structural basis for different ω-agatoxin IVA sensitivities of the P-type and Q-type Cav2.1 channel:
The P/Q-type Cav channels Cav2.1, encoded by CACNA1A and prevalently found in neurons and neuroendocrine cells, play a vital role in the release of neurotransmitters and the regulation of synaptic plasticity and neuronal excitability. Cav2.1 presents a potential therapeutic target for treating neurological disorders, such as epilepsy and migraine. Our work on Cav2.1 channel reveals the molecular mechanism of subtype-specific blockade of Cav2.1 by MVIIC and Aga-IVA, affords the structural interpretation for the distinct responses of the P- and Q-type Cav2.1 channels to Aga-IVA, and provides insight into the development of more efficient and selective modulators for the treatment of neuronal disorders. [Cell Res 34, 455-457 (2024)]
4. Structure of human Cav2.2 channel blocked by the painkiller ziconotide:
The N-type Cav2.2 is essential for neurotransmitter release in both the central and peripheral nervous systems. Cav2.2 also participates in pain signaling, and suppressing its activity represents a strategy for the development of painkiller. Ziconotide, a synthetic peptide found in the venom of a marine snail, specifically blocks the function of Cav2.2 and has been clinically used to treat intractable pain. We determined the cryo-EM structures of Cav2.2 alone and in complex with ziconotide at resolutions of 3.1 Å and 3.0 Å, respectively. From the structures, we observed that ziconotide is nestled in the electronegative cavity surrounding the entrance to the selectivity filter. The high-resolution structure reveals the molecular basis for Cav2.2-specific pore blocking by ziconotide. [Nature 596, 143-147 (2021)]
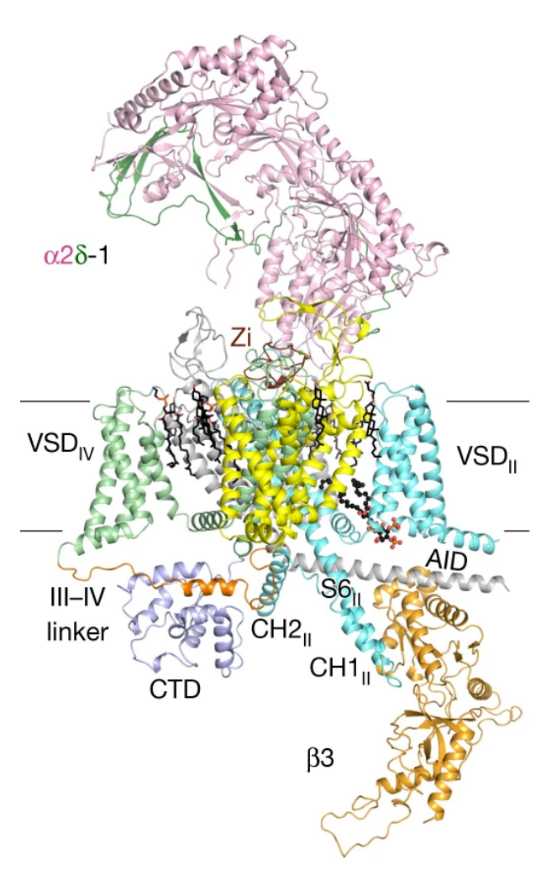
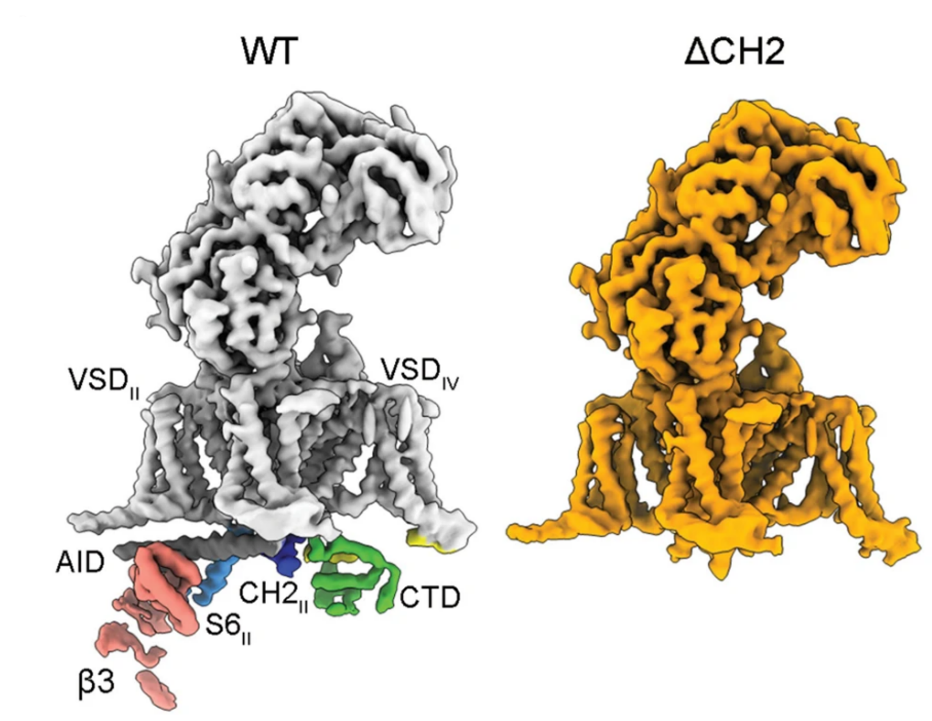
5. Structures of the R-type human Cav2.3 channel reveal conformational crosstalk of the intracellular segments:
The R-type Cav2.3 is widely expressed in neuronal and neuroendocrine cells and represents a potential drug target for pain, seizures, and epilepsy. We determined the cryo-EM structures of Cav2.3 and a mutant named ΔCH2, in which a Cav2-unique cytosolic helix in repeat II (CH2II helix) is deleted, both at a resolution of 3.1 Å. Our structural and electrophysiological characterizations of the WT and ΔCH2 Cav2.3 indicate that the CH2II helix stabilizes the inactivated conformation of the channel by tightening the cytosolic juxtamembrane segments. [Nat. Commun. 13, 7358 (2022)]
6. Cryo-EM structures of apo and antagonist-bound human Cav3.1:
The Cav3 channels are activated at low membrane potentials and are also known as T-type channels due to their transient single-channel conductance. The unique properties of Cav3 channels relate to their specific physiological role in cellular excitability modulation, low-threshold firing, and pacemaker activity. We determined the cryo-EM structures of human Cav3.1 alone and in complex with a highly selective blocker, Z944, at resolutions of 3.3 Å and 3.1 Å, respectively. These structures elucidate the molecular basis for antagonist binding and the distinct channel properties of different Cav subfamilies. [Nature 576, 492-497 (2019)]
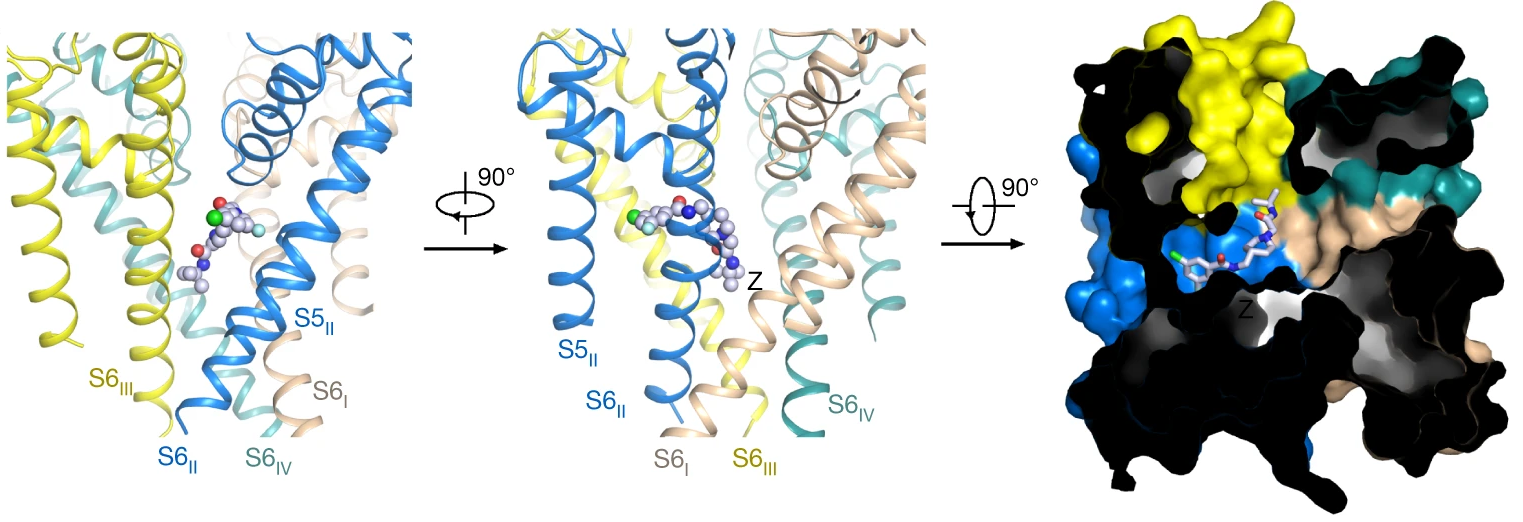
7. Structural basis for human Cav3.2 inhibition by selective antagonists:
The Cav3.2 subtype of T-type calcium channels has been targeted for developing analgesics and anti-epileptics for its role in pain and epilepsy. In our cryo-EM structures of Cav3.2 alone and in complex with four T-type calcium channel selective antagonists (ACT-709478, TTA-A2, TTA-P2 and ML218), we found the four compouds display two binding poses and the fenestration-penetrating mode affords an explanation for the state-dependent activities of these antagonists. Structure-guided mutational analysis identifies several key residues that determine the T-type preference of these drugs. The structures also suggest the role of an endogenous lipid in stabilizing drug binding in the central cavity. [Cell Res 34, 440-450 (2024)]