We combine structural biology, biochemical, and biophysical approaches to study the structure and mechanism of transmembrane transport proteins. We aim to reveal the molecular basis for their physiological and pathophysiological functions.

We combine structural biology, biochemical, and biophysical approaches to study the structure and mechanism of transmembrane transport proteins. We aim to reveal the molecular basis for their physiological and pathophysiological functions.
Glucose is an essential fuel for most living organisms on Earth. Glucose transporters (GLUTs), which belong to the major facilitator superfamily (MFS) of passive transporters, facilitate the diffusion of glucose and other hexoses across the plasma membrane. Among the 14 members of human GLUT family, GLUT1–4 have been rigorously investigated because of their fundamental roles in various physiological and pathophysiological processes.
GLUT1, the first glucose transporter to be cloned, is responsible for glucose uptake into erythrocytes and for transporting glucose across blood–brain barrier. Mutations in GLUT1 can lead to GLUT1 deficiency syndrome (also known as De Vivo disease), which manifests as a broad spectrum of clinical phenotypes including infantile-onset seizure. GLUT2 is highly expressed in pancreatic β-cell, the intestine, kidneys and the liver, and is associated with the Fanconi–Bickel syndrome. GLUT3 is the primary neuronal glucose transporter. The expression levels of GLUT1 and GLUT3 are enhanced in some carcinoma cell lines to increase glucose supply for cancer cells, a phenomenon known as the Warburg effect. GLUT4 is the main glucose transporter in muscles and adipocytes. Unlike other GLUTs, the cellular localization of GLUT4 is regulated by insulin through a complex mechanism, the disruption of which contributes to obesity and type 2 diabetes mellitus.
In the last decade, we have elucidated the structures of glucose transporters in various conformations. These structures together have collectively revealed the alternating access mechanism underlying glucose transport.
Glucose transporters are essential for the metabolism of glucose in cells across a wide range of organisms, from microbes to humans. The importance is exemplified by the disease-related human proteins GLUT1, 2, 3 and 4. Despite rigorous efforts, structural information for GLUT1–4 or their homologues remains largely unknown. We report three related crystal structures of XylE, an Escherichia coli homologue of GLUT1–4, in complex with D-xylose, D-glucose and 6-bromo-6-deoxy-D-glucose, at resolutions of 2.8, 2.9 and 2.6 Å, respectively. The structure features a typical MFS fold of 12 transmembrane segments along with a unique intracellular four-helix domain. XylE was captured in an outward-facing, partly occluded conformation. Most of the key residues responsible for the recognition of D-xylose and D-glucose are invariant in GLUT1–4, suggesting functional and mechanistic conservation. Structure-based modelling of GLUT1–4 allows mapping and interpretation of disease-related mutations. The structural and biochemical information reported here provides an important framework for understanding the mechanisms of glucose transporters and sugar porters in general. [Nature 490, 361-366 (2012)]
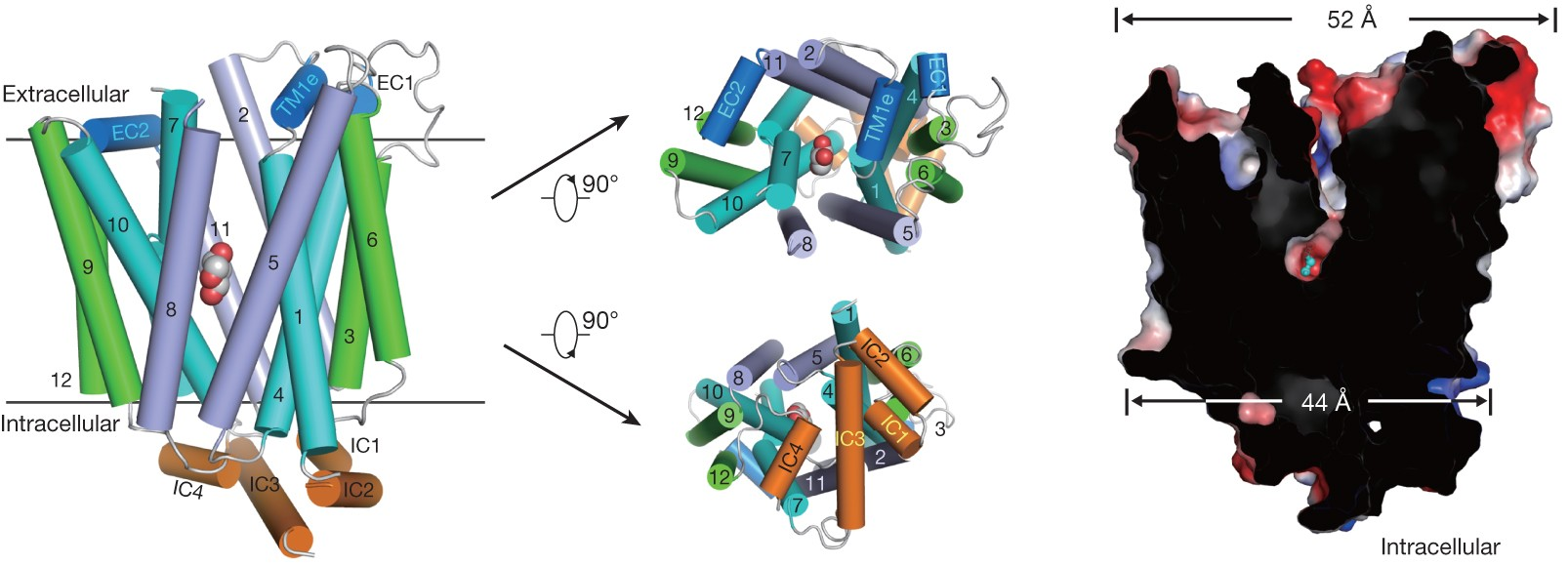
The glucose transporter GLUT1 catalyzes the facilitative diffusion of glucose into erythrocytes and is responsible for supplying glucose to the brain and other organs. Dysfunctional mutations may lead to GLUT1 deficiency syndrome, whereas overexpression of GLUT1 serve as a prognostic indicator for cancer. Despite decades of investigation, the structure of GLUT1 remains unknown. We report the crystal structure of human GLUT1 at a resolution of 3.2 Å. The full-length protein, which exhibits a canonical MFS fold, is captured in an inward-open conformation. This structure enables accurate mapping and potential mechanistic interpretation of disease-associated mutations in GLUT1. Structure-based analysis of these mutations provides insight into the alternating access mechanism of GLUT1 and other members of the sugar porter subfamily. Structural comparison of the uniporter GLUT1 with its bacterial homologue XylE, a proton-coupled xylose symporter, allows the examination of the transport mechanisms of both passive facilitators and active transporters. [Nature 510, 121-125 (2014)]
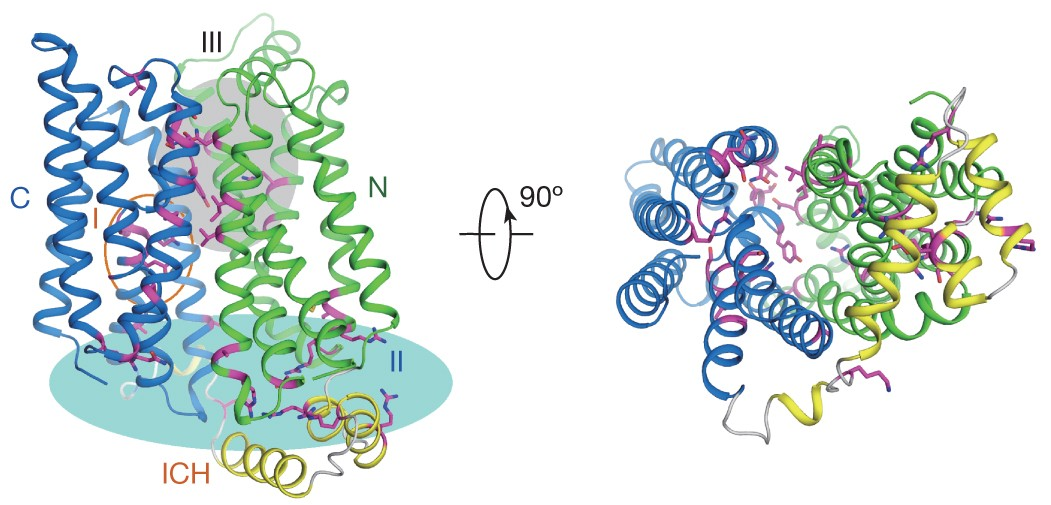
The MFS glucose transporters, exemplified by human GLUT1-4, have been central to the study of solute transport. Using lipidic cubic phase crystallization and microfocus X-ray diffraction, we determined the structure of human GLUT3 in complex with D-glucose at a resolution of 1.5 Å in an outward-occluded conformation. The high-resolution structure allows for the discrimination of both alpha- and beta-anomers of D-glucose. Additionally, two structures of GLUT3 bound to the exofacial inhibitor maltose were obtained at 2.6 Å in the outward-open state and 2.4 Å in the outward-occluded state. In all three structures, the ligands are predominantly coordinated by polar residues from the C-terminal domain. The conformational transition from outward-open to outward-occluded entails a notable local rearrangement of the extracellular portion of transmembrane segment TM7. Comparison of the outward-facing GLUT3 structures with the inward-open GLUT1 provides insights into the alternating access cycle for GLUTs, whereby the C-terminal domain serves as the primary substrate-binding site while the N-terminal domain undergoes rigid-body rotation relative to the C-terminal domain. Our studies provide an important framework for the mechanistic and kinetic understanding of GLUTs and illuminate structure-guided ligand design. [Nature 526, 391-396 (2015)]
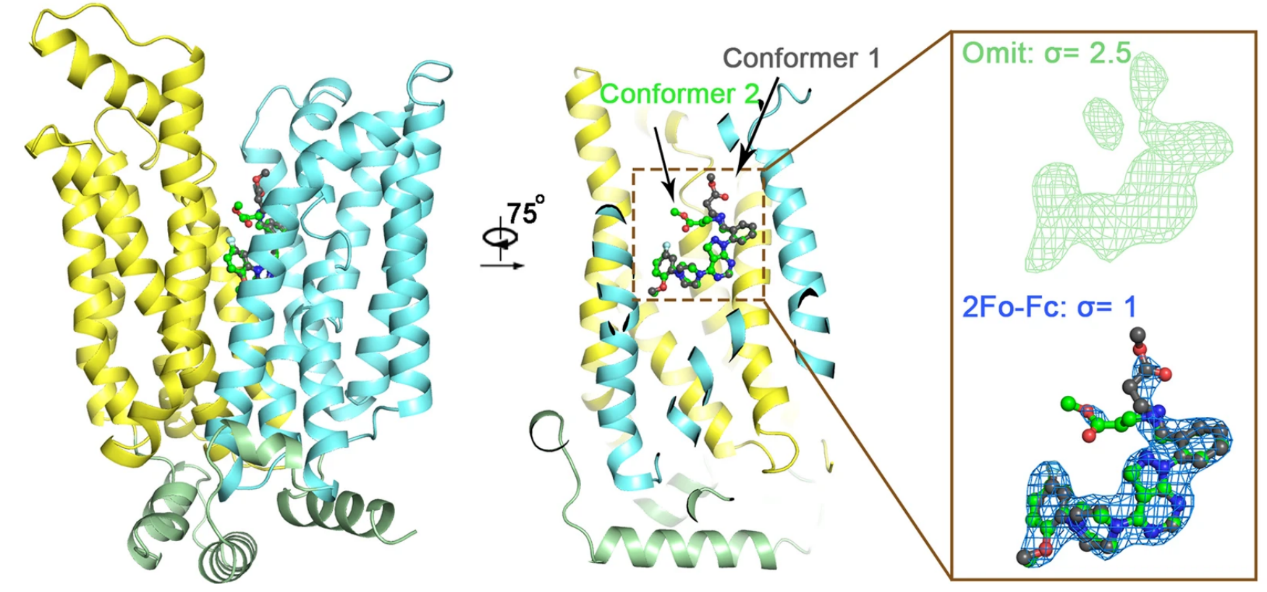
Plasmodium species, the causative agents of malaria, rely on glucose for energy supply during the blood stage. Inhibition of glucose uptake thus represents a potential strategy for the development of antimalarial drugs. We present the crystal structures of PfHT1, the sole hexose transporter in the genome of Plasmodium species, at resolutions of 2.6 Å in complex with D-glucose and 3.7 Å with a moderately selective inhibitor, C3361. Although both structures exhibit occluded conformations, the binding of C3361 induces marked rearrangements that create an additional pocket. This inhibitor-binding-induced pocket offers an opportunity for the rational design of PfHT1-specific inhibitors. Among our designed C3361 derivatives, several exhibited improved inhibition of PfHT1 and cellular potency against P. falciparum, with excellent selectivity for human GLUT1. These findings serve as a proof of concept for the development of next-generation antimalarial chemotherapeutics by simultaneously targeting the orthosteric and allosteric sites of PfHT1. [Cell 183, 258-268 (2020)]
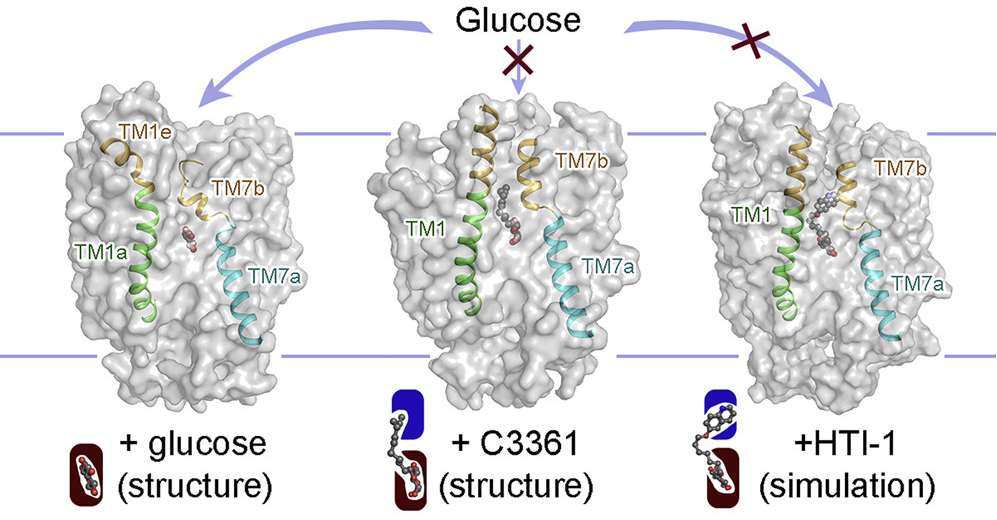
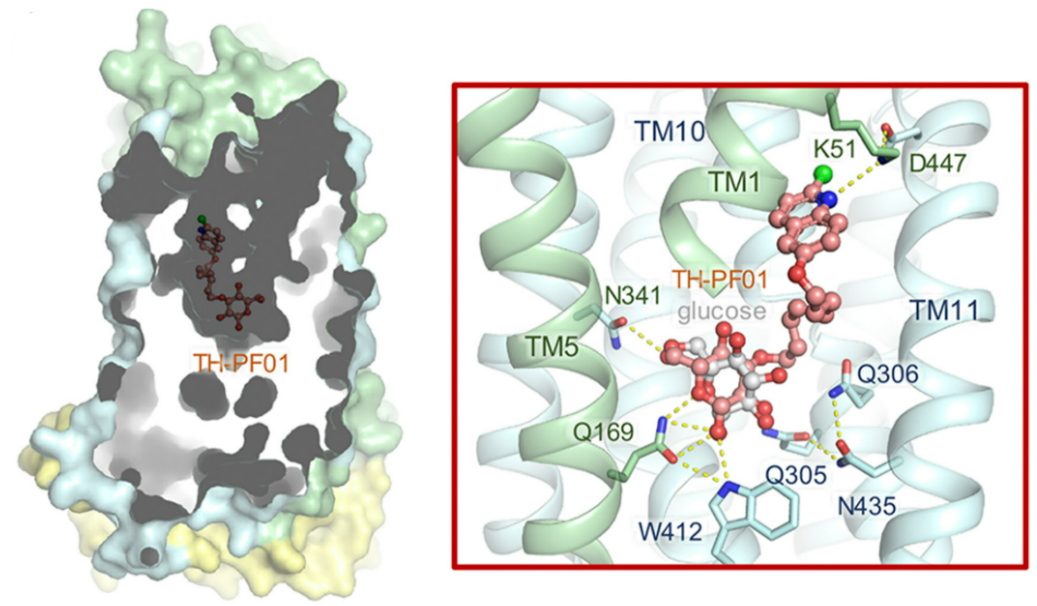
Artemisinin-resistant malaria parasites have emerged and are spreading, posing a significant public health challenge. Therefore, antimalarial drugs with novel mechanisms of action are urgently needed. We exploit a “selective starvation” strategy by inhibiting Plasmodium falciparum hexose transporter 1 (PfHT1), the sole hexose transporter in P. falciparum, over human glucose transporter 1 (hGLUT1), providing an alternative approach to combat multidrug-resistant malaria parasites. The crystal structure of hGLUT3, which shares 80% sequence similarity with hGLUT1, was resolved in complex with C3361, a moderate PfHT1-specific inhibitor, at 2.3 Å resolution. Structural comparison between the present hGLUT3-C3361 and our previously reported PfHT1-C3361 confirmed the unique inhibitor binding-induced pocket in PfHT1. We then designed small molecules to simultaneously block the orthosteric and allosteric pockets of PfHT1. Through extensive structure–activity relationship studies, the TH-PF series was identified as selectively inhibiting PfHT1 over hGLUT1 and showing potency against multiple strains of blood-stage P. falciparum. Our findings provide insights into the next-generation chemotherapeutics employing a paradigm-shifting structure-based design strategy to target both the orthosteric and allosteric sites of a transporter simultaneously. [PNAS 118, e2017749118 (2021)]
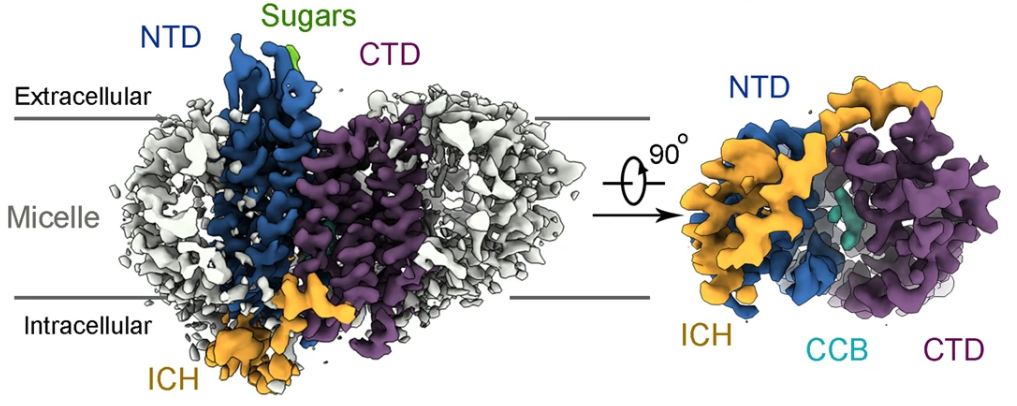
GLUT4 is the primary glucose transporter in adipose and skeletal muscle tissues and its cellular trafficking is regulated by insulin signaling. Failed or reduced plasma membrane localization of GLUT4 is associated with diabetes. We report the cryo-EM structures of human GLUT4 bound to the small molecule inhibitor cytochalasin B (CCB) at resolutions of 3.3 Å in both detergent micelles and lipid nanodiscs. The CCB-bound GLUT4 exhibits an inward-open conformation. Despite the nearly identical conformation of the transmembrane domain to GLUT1, the cryo-EM structure reveals an extracellular glycosylation site and an intracellular helix that is not visible in the crystal structure of GLUT1. The structural study presented here lays a foundation for further mechanistic investigations into the modulation of GLUT4 trafficking. Our methods for cryo-EM analysis of GLUT4 will also facilitate structural determination of many other small solute carriers. [Nat. Commun. 13, 2671 (2022)]
Elevated expression of GLUTs, particularly GLUT1 and GLUT3, is required to fuel the hyperproliferation of cancer cells, making GLUT inhibitors potential anticancer therapeutics. Meanwhile, GLUT inhibitor-conjugated insulin is being explored to mitigate the hypoglycemia side effect of insulin therapy in type 1 diabetes. Reasoning that exofacial inhibitors of GLUT1/3 may be more favorable for therapeutic applications, we report the engineering of a GLUT3 variant, designated GLUT3exo, that can be probed for screening and validating exofacial inhibitors. We identify an exofacial GLUT3 inhibitor, SA47, and elucidate its mode of action by a 2.3 Å resolution crystal structure of SA47-bound GLUT3. Our studies serve as a framework for the discovery of GLUTs exofacial inhibitors for therapeutic development. [Nat. Commun. 13, 2632 (2022)]